PSSM construction
A PSSM was created using the PSI-BLAST PSSM creation protocol as described in the work of Stephen Altschul and colleagues (see 1,2). The 18 validated human, yeast and viral B56 binding peptides (see Validated Instances table) were aligned around their motif and cut to include only their P1-P9 positions. The columns of the alignment at each position are converted to a frequency matrix (Figure 1). Sequences weights were calculated to remove any bias introduced from closely related sequences (though this was not an issue with the currently available set of validated instances). The frequency matrix was weighted using these sequences weights.
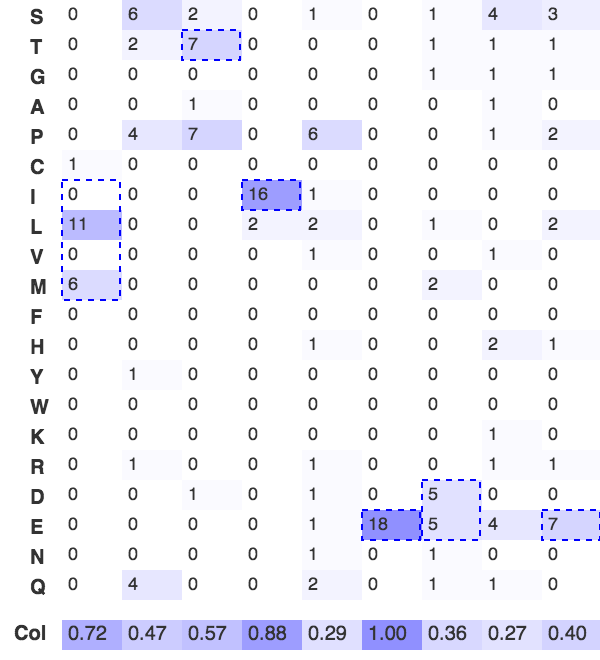
Figure 1. Frequency matrix for the B56-PP2A docking motif based on 18 experimentally validated motifs.
The frequency matrix was altered with pseudocounts to account for the limited number of samples in the validated instance set. The pseudocounts add "expected" data to the matrix for physicochemically similar residues to the observed residues in validated instances at a given position based on the expected probability encoded in the BLOSUM62 matrix. The logic to such a weighting scheme is that we have an observed only a subset of the binding peptides. Residues that physicochemically similar to the observed residues at a position could be permitted as they may also be complementary peptide binding partner at that site in the binding pocket. Finally, scaling of this pseudocount matrix results in a position specific scoring matrix (PSSM)(Figure 2).
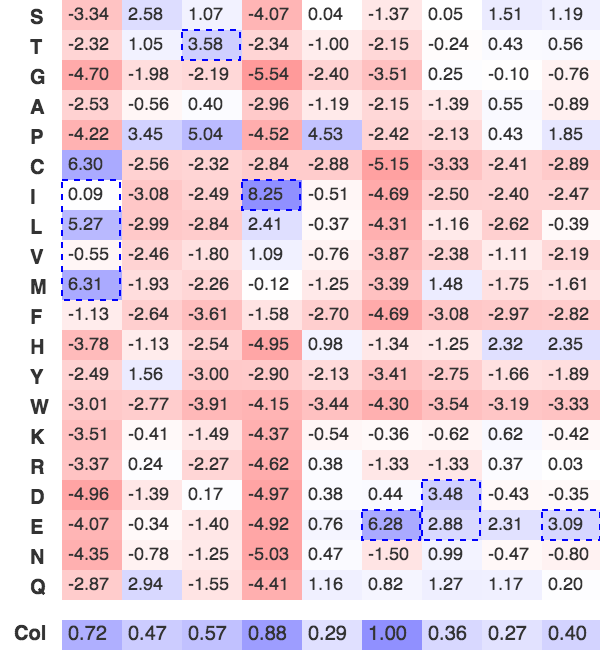
Figure 2. PSSM for the B56-PP2A docking motif based on 18 experimentally validated motifs.
Scoring
Proteins are scored by sliding across the sequence of the protein one residue at a time, taking a peptide of the same length as the PSSM and scoring the peptide. A peptide is scored by taking each position of the peptide, retrieving the PSSM score for the given amino acids at the given position and summing these scores for the complete peptide.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ.
Schäffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF.